Biosimilární léky: co by měl revmatolog vědět
03/2023
MUDr. Hana Ciferská, Ph.D.1; MUDr. Jan Vachek2-3
1Revmatologická klinika 1. LF UK a Revmatologický ústav, Praha
2Klinika nefrologie 1. LF UK a VFN, Praha
3Interní oddělení a hemodialyzační středisko, Klatovská nemocnice, Klatovy
SOUHRN
Rozvoj biotechnologií a genetického inženýrství na konci minulého tisíciletí přinesl přelom v terapii nemocných se zánětlivými revmatickými chorobami, u kterých selhala terapie konvenčními syntetickými chorobu modifikujícími antirevmatickými léky (conventional synthetic disease modifying anti-rheumatic drug csDMARD). Nástup biologické terapie zlepšil prognózu a kvalitu života pacientů, ale přinesl ekonomickou zátěž spojenou s dosud celosvětově vysokou cenou této léčby. Narůstající potřeba biologických chorobu modifikujících antirevmatických léků (biological disease modifying anti-rheumatic drug, bDMARD) a jejich cena vedla po vypršení patentu na originální bDMARD k nástupu biosimilárních léků (biosimilar disease modifying anti-rheumatic drug, bsDMARD). Vzhledem k složité proteinové struktuře nelze bsDMARD považovat za tradiční generika, ale vysoce podobné kopie bDMARD, ze kterých vycházejí. Účinnost, bezpečnost a biosimilarita srovnatelná s originálním bDMARD musí být prokázána prostřednictvím klinických sledování před zavedení bsDMARD do klinické praxe. Předpokládá se, že cenově dostupnější bsDMARD umožní celosvětově širší dostupnost a udržitelnost léčby. Schválení prvního bsDMARD CT-P13 (biosimilární infliximab) se stalo přelomovou událostí, která umožnila rozvoj dalších bsDMARD. Tento článek se zabývá vybranými bsDMARD využívaných v současnosti v terapii zánětlivých revmatických onemocnění v České republice.
Klíčová slova
biologická léčba, biosimilars, infliximab, etanercept, adalimumab, rituximab, revmatoidní artritida, ankylozující spondylitida, psoriatická artritida
SUMMARY
The advent of biotechnology and genetic engineering at the close of the last millennium marked a significant advancement in the treatment of patients with inflammatory rheumatic diseases, particularly when conventional disease-modifying drug therapy (csDMARD) had proven ineffective. The introduction of biological therapies (bDMARDs) has improved patient prognosis and quality of life, yet it has also resulted in an economic burden due to the persistently high cost of these treatments globally. The growing demand for bDMARDs and their associated costs have facilitated the emergence of biosimilars (bsDMARDs) following the expiration of the original bDMARDs patents. Given their complex protein structure, bsDMARDs should not be considered as traditional generics but rather as highly similar replicas of the original bDMARDs. Efficacy, safety, and biosimilarity to the original bDMARD must be demonstrated through clinical monitoring before bsDMARDs can be incorporated into clinical practice. The introduction of more affordable bsDMARDs is expected to expand treatment accessibility and sustainability worldwide. The approval of the first bsDMARD, CT-P13 (biosimilar infliximab), was a pivotal event, paving the way for the development of additional bsDMARDs. This article focuses on selected bsDMARDs currently used in the treatment of inflammatory rheumatic diseases in the Czech Republic.
Key words
biological treatment, biosimilars, infliximab, etanercept, adalimumab, rituximab, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis
Celý článek je dostupný pouze pro předplatitele
Staňte se pravidelným odběratelem našeho časopisu Revue Farmakoterapie...
ÚVOD
Biologické chorobu modifikující antirevmatické léky (biological disease modifying anti-rheumatic drug, bDMARD) výrazně zlepšily mortalitu a morbiditu pacientů se závažnými chronickými autoimunitními revmatickými chorobami. Postupem doby se indikace bDMARD rozšířila na celou řadu revmatologických diagnóz. Nákladnost terapie bDMARD je vyvážena jejich léčebným benefitem, nicméně pro zvýšení počtu léčených nemocných je nutné snížit ekonomickou náročnost léčby. Úspěch bDMARD a vypršení platnosti ochranných patentů umožnilo vývoj biosimilárních chorobu modifikujících antirevmatických léků (biosimilar disease modifying anti-rheumatic drug, bsDMARD). Prvním bsDMARD uvedeným do klinické praxe byl biosimilární infliximab CT-P13 a další následovaly. V poslední dekádě se výrazně zvýšil počet dostupných a vyvíjených bsDMARD. Cena bsDMARD je nižší, ale ne úplně nízká vzhledem k nákladům spojeným s jejich výrobou a nutností klinických studií hodnotících jejich farmakokinetiku, farmakodynamiku, účinnost a bezpečnost vzhledem ke srovnávanému originálnímu bDMARD. Složitá struktura proteinů bDMARD ovlivňuje jejich biologickou dostupnost a tím i účinnost. Primární struktura proteinové molekuly léku je daná sekvencí bází, kterou lze relativně snadno zkopírovat, ale sekundární a terciální a posttranslační modifikace jsou závislé na produkující buněčné linii a podmínkách, ve kterých je udržována. Celý proces replikace může být negativně ovlivněn nedokonalou separací výsledného produktu danou celou řadou faktorů a může tak zvyšovat riziko potenciálních imunologicky zprostředkovaných nežádoucích účinků. Technologicky zatím nelze zajistit naprosto identickou molekulární kopii bDMARD, ale lze dosáhnout velmi podobné struktury bsDMARD vzhledem k originálnímu léku.1,2 Světová zdravotnická organizace (World Health Organization, WHO) definovala bsDMARD jako biologické léky, které by se měly shodovat kvalitou, bezpečností a účinkem s již licencovanými referenčními biologickými léky, které již byly schváleny regulační lékovou agenturou daného státu.3,4 Schválení bsDMARD a jejich zavedení do klinické praxe je podmíněno úspěšným splněním podmínek analytických, preklinických a klinických studií. Nejstarší a nejpočetnější skupina bDMARD využívaných v revmatologii j e skupina inhibitorů tumor nekrotizujícího faktoru α (TNF – α), z nichž u většiny originálních přípravku vypršel ochranný patent (tab. 1 a 2).1 Prvním bsDMARD registrovaným v České republice pro revmatická a muskuloskeletální onemocnění byl biosimilární infliximab CT-P13 (Remsima [Celltrion Healthcare], Inflectra [Hospira Zagreb]), jehož referenčním bDMARD byl infliximab (Remicade, MSD). V současnosti jsou v České republice k dispozici bsDMARD infliximabu, etanerceptu a adalimumabu.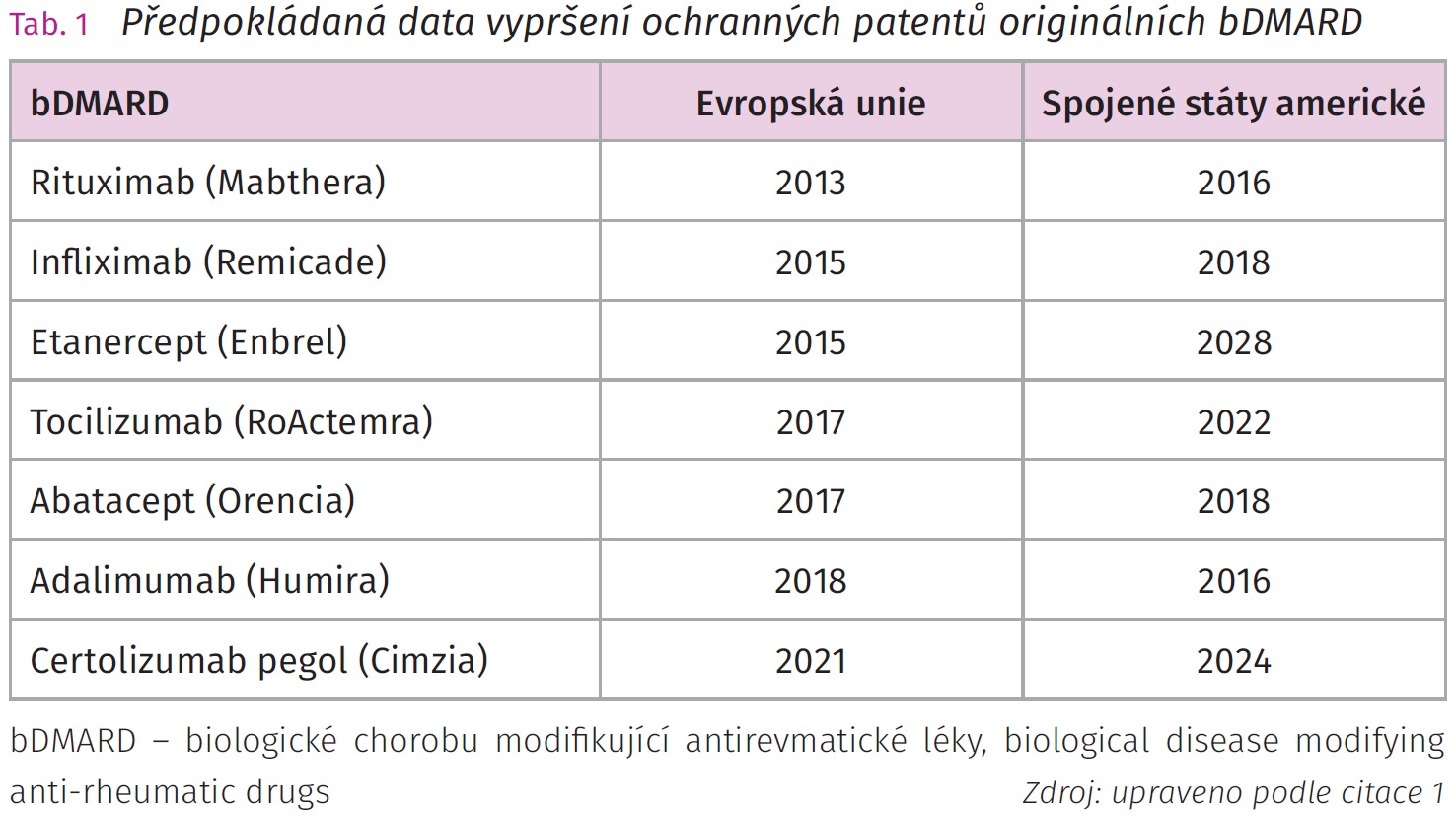
Dalším přípravkem, jehož biosimilární forma je využívána v léčbě revmatických chorob, je protilátka proti povrchovému receptoru CD20 – rituximab. Předpokládá se zavedení dalších bsDMARD v okamžiku vypršení ochranných patentů originálních bDMARD. Indikace podání bDMARD mají svá pevná pravidla podle ustanovení České revmatologické společnosti České lékařské společnosti J. E. Purkyně, která vycházejí z nadnárodních doporučení jak evropských podle European Alliance of Associations for Rheumatology (EULAR), tak amerických podle American College of Rheumatology (ACR).6,7 Indikace podávání bsDMARD se mohou oproti originálnímu bDMARD lišit, což je dáno schvalovacím procesem a terapeutickými cíli klinických sledování.
Dostupnost bsDMARD v České republice
Prvním registrovaným bsDMARD v České republice byl CT-P13 (biosimilární infliximab) a postupně následovaly další molekuly od různých farmaceutických firem. Vypršení ochranných patentů umožnilo produkci bsDMARD vycházejících z originálních molekul. Inhibitory TNF – α (infliximab, adalimumab, etanercept) a rituximab (protilátka anti-CD20) jsou již dostupné ve své biosimilární podobě vycházející z originálního bDMARD. Originální bDMARD je jen jeden, zatímco je celá řada bsDMARD, které z něho vycházejí. Před zavedením do klinické praxe musí každý bsDMARD splnit podmínky registrace (tab. 2).8
INFLIXIMAB
Originální infliximab (Remicade) je inhibitor TNF – α a byl jedním z prvních bDMARD z této skupiny, kterému vypršel ochranný patent a stal se jedním z prvních referenčních bDMARD pro své bsDMARD (CT-P13, SB2, GP1111).
Charakteristika a dávkování infliximabu a jeho biosimilárních variant
Infliximab je monoklonální chimérická protilátka složená z lidského imunoglobulinu G1k (IgG1K) a z myšího antigen vázajícího fragmentu imunoglobulinu (antigen-binding fragment, Fab) se schopností vazby na transmembránovou i solubilní formu TNF – α, ale neváže se na TNF- β. Lze ho podávat intravenózně i subkutánně. Podle revmatologické indikace se liší dávkovací schéma. Intravenózní podávání infliximabu u revmatoidní artritidy (RA) v dávce 3 mg/kg v týdnech 0, 2, 4 a následně v intervalu osmi týdnů. V indikaci léčby ankylozující spondylitidy (AS) a psoriatické artritidy (PsA) je doporučováno odlišné dávkování a intervaly podání, kde je iniciální dávka 5 mg/kg opakovaná po dvou a čtyřech týdnech následně v intervalech šest až osm týdnů.10 Stejné dávkovací schéma je zachováno i u biosimilárních infliximabů (Remsima, Inflectra, Flixabi, Zessly).9-13 Biosimilární infliximab CT-P13 (Remsima) lze podávat i subkutánně.10 U revmatoidní artritidy je nutné zahájit nasycovací dávkou, která může být intravenózní nebo subkutánní. V případě subkutánního podávání je podáváno 120 mg v týdnu 1, 2, 3, 4 a následně pak každé dva týdny. V případě intravenózní nasycovací dávky se přechází na subkutánní formu za čtyři týdny po druhém intravenózním podání. U diagnózy AS a PsA je nasycovací dávka intravenózní ve formě dvou infuzí v dávce infliximabu 5 mg/kg podaných v rozmezí dvou týdnů následované za čtyři týdny subkutánní injekcí, která je pak podávána každé dva týdny (tab. 3).10
CT-P13 – Remsima/Inflectra
Přípravek CT-P13 byl prvním biosimilárním inhibitorem TNF – α, který byl uveden do klinické praxe. Jihokorejská firma Celltrion ho uvedla na evropský trh pod dvěma názvy Remsima a Inflectra v indikaci RA a AS. Dvě velké randomizované dvojitě zaslepené studie fáze III PLANETRA a PLANETAS poskytly dostatek podkladů pro následnou registraci CT-P13 (tab. 4).
Intravenózní infliximab CT-P13 – Remsima/Inflectra
Terapeutickou ekvivalenci přípravku CT-P13 ověřily dvě klinické studie fáze III. Studie PLANETRA14 byla randomizovaná dvojitě zaslepená multicentrická studie fáze III. srovnávající bezpečnost a účinnost CT-P13 s referenčním infliximabem u pacientů s RA (CT-P13 n = 302 a infliximab n = 304), u kterých došlo k selhání terapie methotrexátem (MTX). Bazální terapie MTX byla ponechána u obou ramen studie. Studijní medikace byla podávána intravenózně v dávce 3 mg/kg podle obvyklého schématu pro infliximab. Bylo dosaženo primárního cíle, tj. 20% zlepšení podle American College of Rheumatology (ACR20), ve 30. týdnu.

Mezi sekundární cíle patřilo zhodnocení klinické odpovědi podle celé řady skórovacích indexů zaměřených nejen na klinický stav nemocného, ale také na kvalitu života: podle ACR50 a ACR70, prostřednictvím kritérií EULAR, skóre aktivity nemoci podle hodnocení 28 kloubů (Disease Activity Score, DAS28) a dotazníku kvality života (Medical Outcomes Study Short-Form Health Survey, SF-36). Přípravek CT-P13 prokázal terapeutickou ekvivalenci s referenčním infliximabem, neboť hodnota 95% CI pro rozdíl účinnosti klinické odpovědi podle kritéria ACR20 byla v intervalu ± 15 %. Terapeutické odpovědi ACR20 bylo dosaženo ve 30. týdnu u 60,9 % pacientů s CT-P13 a u 58,6 % pacientů s infliximabem (95% CI 6-10 %). Stran výskytu nežádoucích účinků se obě skupiny rámcově nelišily (CT-P13 35,2 %; referenční infliximab 35,9 %). Farmakologické vlastnosti včetně imunogenity CT-P13 se rámcově nelišily od referenčního infliximabu. Maximální sérová koncentrace (Cmax) obou studijních přípravků byla sledována pro jedno až šest podání a dosahovala průměrně hodnot 83,9-111,9 µg/ml u CT-P13 a u infliximabu pak 83,8-105,1 µg/ml. Přítomnost protilátek proti infliximabu byla zaznamenána u 48,4 % v terapii CT-P13 a u 48,2 % infliximabu.14 Studie PLANETAS15 byla randomizovaná dvojitě zaslepená multicentric - ká studie fáze III srovnávající CT-P13 s referenčním infliximabem v populaci pacientů s AS (CT-P13 n = 125 a infliximab n = 125), u kterých došlo k selhání předchozí konvenční terapie. Přípravky CT-P13 i infliximab byly podávány intravenózně v dávce 5 mg/kg v obou ramenech studie podle obvyklého schématu v indikaci AS. Primární cíle studie byly plocha pod křivkou koncentrace léčiva v krvi (area under the curve, AUC) a maximální plazmatická koncentrace v ustáleném stavu (Cmax,ss) mezi 22. a 30. týdnem klinického sledování. Geometrický průměr hodnot AUC byl 32 765,8 µgh/ml u CT-P13 a 31 359,3 µgh/ml pro infliximab. Geometrický průměr Cmax,ss byl 147,0 pg/ml pro CT-P-13 a 144,8 µg/ml u infliximabu. Mezi sekundární cíle patřilo porovnání farmakokinetických a farmakodynamických vlastností CT-P13 s referenčním infliximabem včetně jeho účinnosti hodnocené pomocí 20 % a 40% zlepšení podle skóre ASAS (Assessment in Ankylosing Spondylitis international working group, ASAS20 a ASAS40) a bezpečnosti.

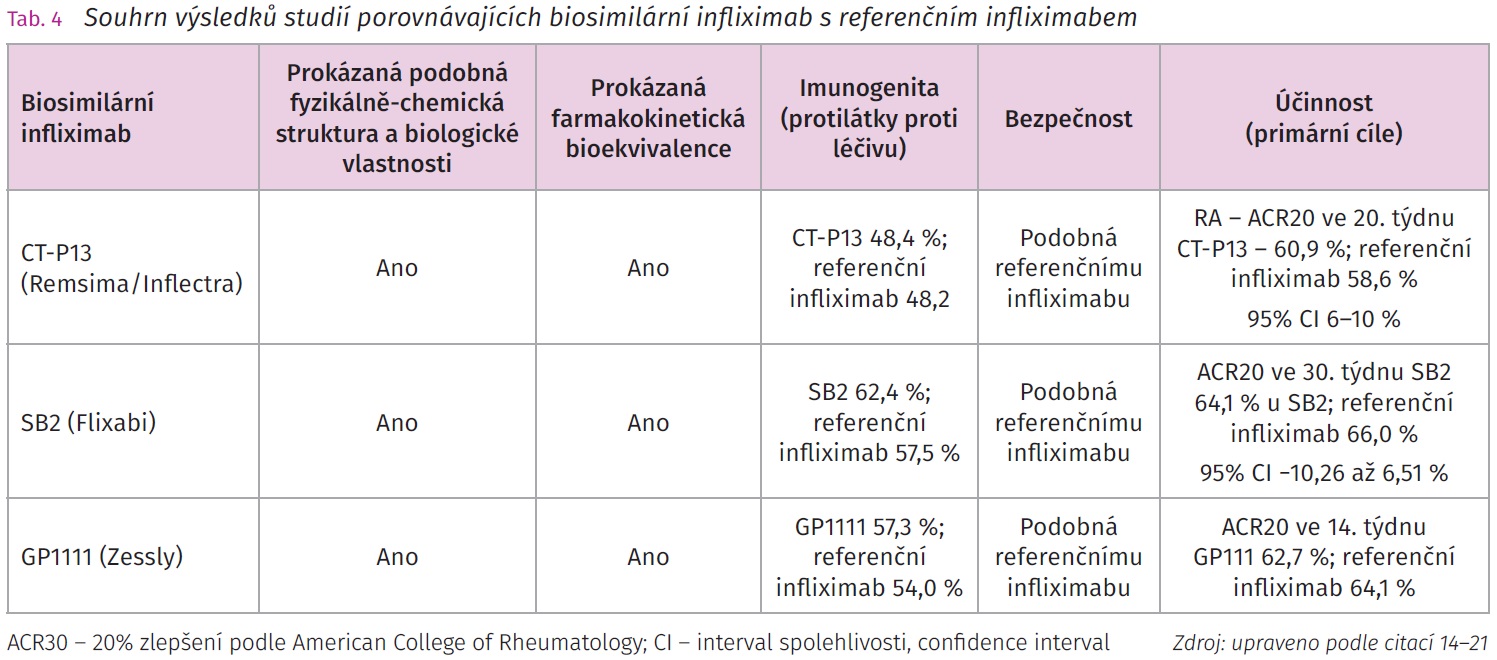
Ve 30. týdnu bylo dosaženo ASAS20 a ASAS40: CT-P13 (70,5 % a 51,8 %) a infliximab (72,4 % a 47,4 %). Nebyl zaznamenán výrazný rozdíl ve výskytu nežádoucích příhod mezi CT-P13 (64,8 %) a referenčním infliximabem (63,9 %). Protilátky proti studijní medikaci (anti-drug antibodies, ADA) byly zaznamenány u 27,4 % pacientů dostávajících CT-P13 a u 22,5 % pacientů s infliximabem. Dvouletá extenze této studie potvrdilo účinnost a bezpečnost CT-P13 ve srovnání s referenčním infliximabem.15
Subkutánní infliximab CT-P13 – Remsima
Subkutánní podávání snižuje časovou a personální náročnost proti intravenózní aplikaci. Umožňuje pacientovi větší autonomii s méně častými kontrolami ve zdravotnickém zařízení.
Studie NCT0314724816 byla dvojitě zaslepená studie fáze I/III porovnávající subkutánní a intravenózní formu CT-P13 v populaci pacientů s RA (n = 343), u kterých došlo k selhání konvenční terapie MTX. Úvodem studie před randomizací byl podán všem pacientům CT-P13 3 mg/kg i.v. v týdnu 0 a 2, následně byli v týdnu 6 pacienti rozděleni v poměru 1: 1 do skupin podle podávané studijní medikace (CT-P13 podávaný subkutánně v dávce 120 mg 1x za dva týdny, n = 167 a CT-P13 podávaný intravenózně v dávce 3 mg/kg každých osm týdnů, n = 176) v této medikaci pokračovali do týdne 22 a od týdne 30 užívali všichni pacienti CT-P13 až do ukončení prodloužené fáze studie v týdnu 64 v subkutánní formě. Primární cíl studie bylo dosažení srovnatelné terapeutické odpovědi hodnocené podle DAS28 vypočteného podle koncentrace C-reaktivního proteinu (DAS28-CRP) v týdnu 22, který byl dosažen. Průměrný pokles DAS28-CRP proti výchozí hodnotě byl v týdnu 22 pro CT-P13 s.c. 2,21 a 1,94 pro CT-P13 i.v., tj. rozdíl 0,27 (95% CI 0,02-0,52). Výskyt nežádoucích příhod vycházel nepatrně lépe pro subkutánní formu (54,8 %) oproti intravenózní formě CT-P13 (66,9 %). Byla prokázána srovnatelná účinnost, bezpečnost a imunogenita s.c. a i.v. CT-P13.16
Metaanalýza zahrnující data dvou randomizovaných studií porovnávajících účinnost a bezpečnost intravenózní a subkutánní formy CT-P13 prokázala, že subkutánní forma CT-P13 je spojena s lepší terapeutickou odpovědí hodnocenou podle DAS28-CRP, ACR20/50/70, klinického indexu aktivity onemocnění (Clinical Disease Activity Index, CDAI) a zjednodušeného indexu aktivity onemocnění (Simplified Disease Activity Index, SDAI).17
SB2 – Flixabi
Přípravek SB2 – Flixabi je biosimilární infliximab vyvinutý firmou Samsung Bioepis / Biogen. Jedná se o intravenózně podávaný bsDMARD využívaný v revmatologických indikacích (tab. 3).
Studie NCT01936181, EudraCT 2012-005733-37 byla randomizovaná dvojitě zaslepená mezinárodní multicentrická studie fáze III s paralelními skupinami nemocných se středně těžkou až těžkou RA (n = 584) se selháním předchozí terapie MTX. Pacienti byli randomizováni podle podávané studijní medikace SB2 (n = 291; analyzováno n = 290) a referenční infliximab (n = 293). Hlavní část studie trvala 54 týdnů s navazující 24týdenní extenzí. U části pacientů byl proveden switch mezi SB2 a referenčním infliximabem. Studijní medikace byla podávána podle obvyklého schématu infliximabu v terapii RA v dávce 3 mg/kg. Primární cíl představovalo dosažení terapeutické odpovědi ACR20 v týdnu 30 s požadovanou ekvivalencí hodnot 95 % CI v rozmezí ± 15 %. Odpovědi ACR20 bylo v týdnu 30 u SB2 dosaženo v 64,1 % oproti 66,0 % u infliximabu (95% CI -10,26 až 6,51), což bylo v rámci předem definovaného rozpětí ekvivalence. Terapeutická ekvivalence obou studijních přípravků byla potvrzena dosažením sekundárních cílů v indexech DAS28, ACR50 a ACR70. V týdnu 54 skupiny s SB2 a s referenčním infliximabem vykazovaly obdobnou terapeutickou odpověď hodnocenou dosažením ACR20 (50,7 % a 52,6 %), ACR50 (32,1 % a 29,7 %) a ACR70 (18,3 % a 17,7 %). Radiografická progrese byla srovnatelná mezi oběma skupinami. Výskyt nežádoucích příhod byl obdobný v obou skupinách (57,6 % a 58,0 %), stejně jako výskyt protilátek proti infliximabu do 30. týdne (55,1 % a 49,7 %). V týdnu 30 byl výskyt nežádoucích příhod vyvolaných léčbou srovnatelný mezi SB2 a referenčním infliximabem (57,6 % a 58,0 %). Farmakologické vlastnosti SB2 a referenčního infliximabu byly srovnatelné. V týdnu 54 nebyl zjištěn statisticky významný rozdíl v přítomnosti autoprotilátek u skupin s SB2 a referenčním infliximabem (62,4 a 57,5 %).18 V týdnu 54 nebyl zjištěn významný rozdíl ve výskytu nežádoucích příhod vyvolaných léčbou po léčbě SB2 a referenčním infliximabem (61,7 % a 65,2 %). Další část studie následovala v 54. týdnu, kdy došlo k rerandomizaci pacientů, a to buď v pokračování léčby infliximabem (n = 101), nebo byli pacienti převedeni na SB2 (n = 94). Ti, kteří byli léčeni SB2 (n = 201), pokračovali v léčbě. Vyhodnocení dat v týdnu 78 prokázalo, že dosažení ACR20, ACR50 a ACR70 bylo srovnatelné ve všech léčebných skupinách.19 Terapeutická odpověď a bezpečnost byla konstantní i v týdnu 78. Protilátky proti léčivu byly dokumentovány u 45,7-53,6 % pacientů; mezi pacienty, kteří nevykazovali přítomnost protilátek do 54. týdne, byly nově vzniklé ADA zaznamenány u 14,6 % pacientů, kteří přešli z referenčního infliximabu na SB2, u 14,9 % pacientů, kteří pokračovali v léčbě referenčním infliximabem, a u 14,1 % pacientů, kteří pokračovali v léčbě SB2. V 78. týdnu byly dokumentovány nežádoucí příhody vyvolané léčbou u 36,2 % pacientů, kteří přešli z referenčního infliximabu na SB2, u 35,6 % pacientů, kteří pokračovali v léčbě referenčním infliximabem a u 40,3 % pacientů, kteří pokračovali v léčbě SB2.19
PF-06438179/GP1111 – Zessly
PF-06438179/GP1111 – Zessly je biosimilární infliximab firmy Pfizer-Sandoz.13
Studie REFLECTIONS B537-02 byla randomizovaná multicentrická dvojitě zaslepená studie fáze III srovnávající terapeutické, farmakologické a bezpečnostní vlastnosti PF-06438179/GP1111 (n = 324) s referenčním infliximabem (n = 326) v populaci pacientů se středně těžkou až těžkou RA s anamnézou selhání konvenční terapie MTX. Oba infliximaby, jak biosimilární, tak referenční, byly podávány podle obvyklého schématu u RA (3 mg/kg v týdnu 0, 2, 6 a následně každých osm týdnů). V týdnu 14 dosáhlo odpovědi ACR20 62,7 % pacientů s PF-06438179/GP1111 a 64,1 % s infliximabem, tato odpověď byla konstantní i v týdnu 30, přičemž rozdíl v odpovědi ACR20 se pohyboval v rozmezí od -5,81 do 0,83 %. Incidence nežádoucích příhod v průběhu studie byla 57,3 % u PF-06438179/GP1111 oproti 54,0 % u infliximabu. Výskyt ADA byl 48,6 % u PF-06438179/GP1111 a 51,2 % u referenčního infliximabu v týdnu 30. Přípravek PF-06438179/GP1111 prokázal bioekvivalenci a srovnatelnou účinnost s referenčním infliximabem v průběhu celého klinického sledování.20'21
ETANERCEPT
Originální etanercept (Enbrel) se stal referenční molekulou pro své biosimilární varianty. Etanercept je unikátní ve skupině inhibitorů TNF – α svou strukturou a mechanismem účinku.
Charakteristika a dávkování etanerceptu a jeho biosimilárních variant
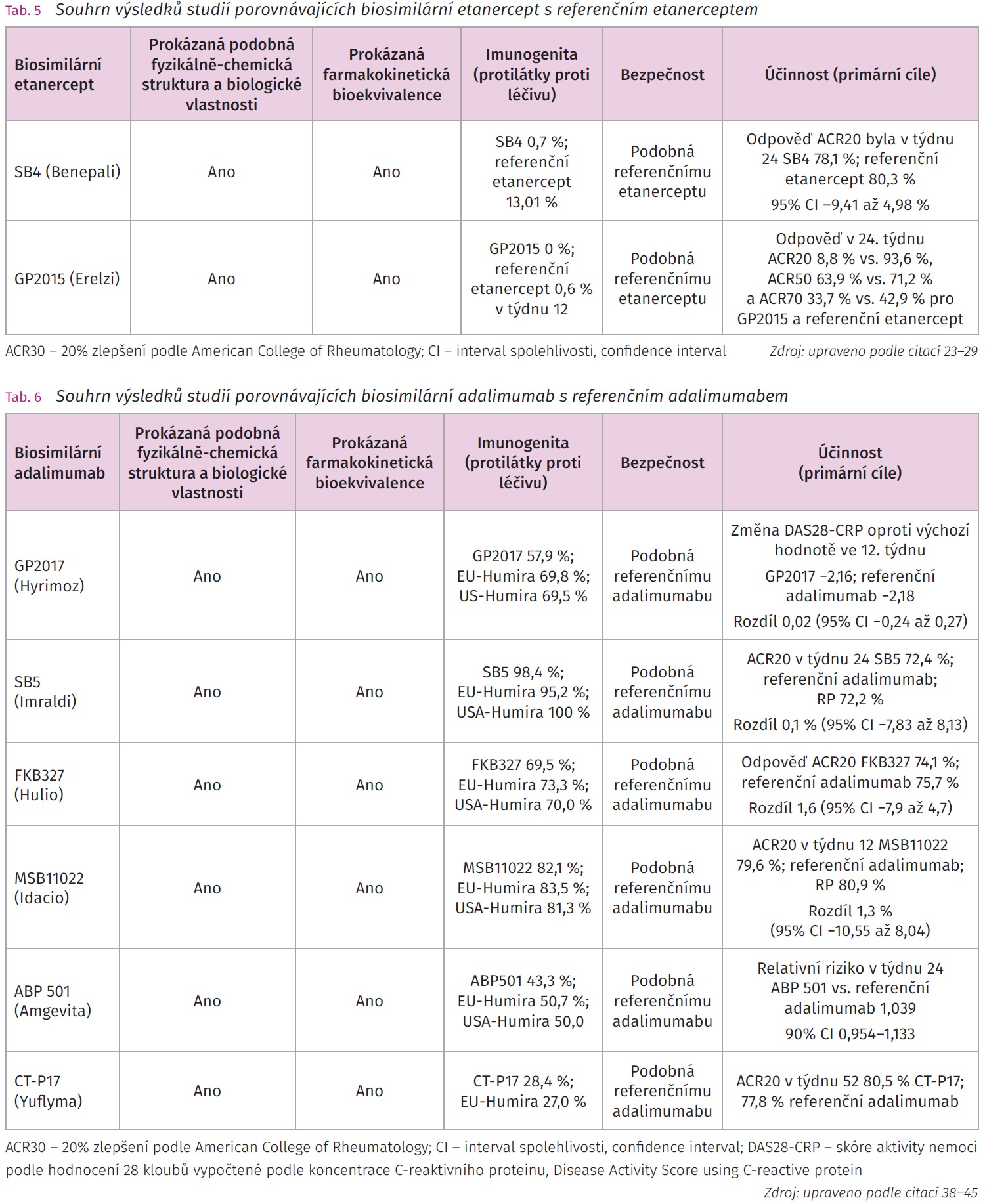
Etanercept je fúzní protein složený z oblasti Fc (fragment crystallizable) lidského IgG a z extracelulární domény receptoru p75 pro TNF-a působící jako kompetitivní inhibitor vazby TNF-a na jeho buněčné receptory. Etanercept se podává subkutánně v dávce 50 mg 1x týdně u dospělé populace k léčbě RA, JIA, PsA, AS a neradiografické axiální spondylartritidy (nr-axSpA).22 Stejné dávkovací schéma je zachováno i u biosimilárních etanerceptů (Benepali, Erelzi) (tab. 5).23,24
SB4 – Benepali
Přípravek SB4 – Benepali je biosimilární etanercept vyvinutý firmou Samsung Bioepis / Biogen.
Studie NCT01895309, EudraCT2012- -005026-30 byla randomizovaná dvojitě zaslepená multicentrické studie fáze III cílená na pacienty se středně těžkou a těžkou RA, u kterých došlo k selhání předchozí terapie MTX. Randomizace byla provedena 1: 1 pro SB4 (n = 299) a referenční etanercept (n = 297). Studijní medikace byla podávána subkutánně 1x týdně v dávce 50 mg. Primárního cíle ACR20 bylo dosaženo v týdnu 24 u SB4 (78,1 %) i u referenčního etanerceptu (80,3 %) (95 % CI -9,41 až 4,98 %), což splnilo předpoklad bioekvivalence obou přípravků. Terapeutická odpověď se udržela konstantní i v týdnu 54 pro oba přípravky: SB4 80,8 % a referenční etanercept 81,5 %. Výskyt nežádoucích účinků byl u obou skupin srovnatelný: SB4 55,2 % a referenční etanercept 58,2 %. Protilátky proti studijní medikaci byly detekovány pouze u 0,7 % pacientů s SB4 oproti 13,1 % u referenčního etanerceptu.25
Na základní studii navazovala otevřená extenze s prodloužením do týdne 100, do které vstoupilo celkem 245 pacientů. Pacienti užívající SB4 v léčbě pokračovali (n = 126) a 119 pacientů přešlo z referenčního etanerceptu na SB4. Terapeutická odpověď hodnocená podle dosažení ACR20 byla v týdnu 100 u první skupiny 77,9 % a 79,1 % u druhé. Ostatní výsledky účinnosti, včetně radiografické progrese, byly u obou skupin rovněž srovnatelné. Přechod z etanerceptu na SB4 nevedl k žádnému zvýšení imunogenity.25-27
GP2015 – Erelzi
Přípravek GP2015 – Erelzi je biosimilární etanercept vyvinutý firmou Sandoz.24 Schválení GP2015 bylo umožněno nejen extrapolací dat získaných ze studie EGALITY, která srovnávala GP2015 s referenčním etanerceptem u 531 pacientů s plakovou psoriázou, ale i studii EQUIRA u pacientů s RA (tab. 5).
Studie EQUIRA byla dvojitě zaslepená placebem kontrolovaná studie fáze III zaměřená na populaci pacientů se středně těžkou až těžkou RA (n = 379), u kterých selhala konvenční terapie csDMARD. Cílem studie bylo prokázat srovnatelnou účinnost, bezpečnost a imunogenitu GP2015 (n = 181) s referenčním etanerceptem (Enbrel) (n = 172) při bazální terapii MTX. Primárních cílů bylo dosaženo v týdnu 24 s terapeutickou odpovědi ACR20 88,8 % vs. 93,6 %, ACR50 63,9 % vs. 71,2 % a ACR70 33,7 % vs. 42,9 % pro GP2015 a referenční etanercept. Rozdíl mezi odpověďmi hodnocenými podle DAS28-CRP byl v předem specifikovaném rozmezí ekvivalence -0,6 až 0,6. Výskyt nežádoucích účinků v průběhu 24týdenního sledování byl u GP2015 (43,5 %) a referenčního etanerceptu (49,5 %) srovnatelný. U žádného z pacientů léčeného přípravkem GP2015 nebyly detekovány ADA, zatímco ve skupině s referenčním etanerceptem bylo ve 4. a 12. týdnu pozitivních 1,6 % a 0,6 % pacientů.28 Ve 24. týdnu byli pacienti s dobrou terapeutickou odpovědí převedeni na léčbu GP2015, ve které pokračovali do 48. týdne. Průměrná změna DAS28-CRP proti výchozímu stavu do 48. týdne byla u obou skupin, tj. u pacientů, kteří užívali GP2015 od počátku studie (2,90 [0,12], n = 148) a u pacientů, kteří přešli z referenčního etanerceptu na GP2015 ve 24. týdnu studie (2,78 [0,13], n = 131), srovnatelná. Srovnatelných výsledků bylo dosaženo i u dalších parametrů ACR20/50/70. Nežádoucí příhody se vyskytovaly u 42,9 % pacientů s GP2015 a u 38,0 % pacientů, kteří přešli z referenčního etanerceptu na GP2015. Výsledky studie EQUIRA prokázaly, že přechod pacientů z referenčního etanerceptu na biosimilární etanercept GP201 neovlivnil účinnost a bezpečnost podávané medikace u pacientů s RA.29
ADALIMUMAB
Originální adalimumab (Humira) je inhibitor TNF – α, ze kterého vychází celá řada biosimilárních variant, a co se týče jejích zástupců, je nejpočetnější.
Charakteristika a dávkování adalimumabu a jeho biosimilárních variant
Adalimumab je plně humánní monoklonální protilátka třídy IgG1 se specifickou vazbou na TNF – α, která neutralizuje prostřednictvím blokády jeho interakce s receptory na povrchu buněk (p55 a p75). Adalimumab se podává subkutánně v intervalu 14 dní v kombinaci s MTX. Originálním adalimumabem, ke kterému jsou vztaženy jeho biosimilární varianty, je Humira.30 Stejné dávkovací schéma je zachováno i u biosimilárních etanerceptů (Hyrimoz, Imraldi, Hulio, Idacio, Amgevita, Yuflyma, Hukyndra). AVT02 (Hukyndra) je příkladem biosimilárního adalimumabu, u kterého došlo k extrapolaci dat studie fáze III zaměřené na populaci pacientů s plakovou psoriázou, což umožnilo schválení a registraci AVT02 i v revmatologických indikacích (tab. 6).31-37
GP2017 – Hyrimoz
Přípravek GP2017 – Hyrimoz byl vyvinut firmou Sandoz a uveden do klinické praxe byl po splnění potřebných požadavků.
Studie ADMYRA byla dvojitě zaslepená randomizovaná studie fáze III zaměřená na pacienty se středně těžkou až těžkou RA (n = 353), u kterých došlo k selhání předchozí terapie csDMARD. Cílem bylo prokázat ekvivalenci v účinnosti a bezpečnosti biosimilárního adalimumabu GP2017 (n = 177) a referenčního adalimumabu (Humira) (n = 176). Terapeutická odpověď byla hodnocena změnou skóre DAS28-CRP i dalšími ukazateli v týdnu 12 oproti počátku.
Studijní medikace byla podávána ve 14denním intervalu, následně v týdnu 24 po vyhodnocení terapeutické odpovědi podle předem daných parametrů pacienti s léčbou GP2017 pokračovali (n = 159) a pacienti s léčbou referenčním adalimumabem byli převedeni na GP2017 (n = 166) po dobu 46 týdnů. Změna DAS28-CRP ve 12. týdnu proti počátku byla obdobná u GP2017 (-2,16) i u referenčního adalimumabu (-2,18) (změna 0,02 [95 % CI -0,24 až 0,27]), což bylo v rozmezí předem daného rozpětí ekvivalence ± 0,6. V druhé fázi studie byla rovněž terapeutická odpověď podle DAS28-CRP konstantní ve všech skupinách. Výskyt nežádoucích příhod byl srovnatelný u obou skupin. Protilátky proti podávané medikaci byly na počátku 48. týdne zjištěny u 24,2 % pacientů s GP2017 a u 25,6 % pacientů s referenčním adalimumabem.38-40
SB5 – Imraldi
Biosimilární adalimumab SB5 – Imraldi vyvinula firma Samsung Bioepis / Biogen.
Studie NCT02167139 byla randomizovaná dvojitě zaslepená studie fáze III srovnávající SB5 (n = 271) a referenčním adalimumabem (Humira) (n = 273) u pacientů se středně těžkou až těžkou RA se selháním terapie MTX. Primárním cílem bylo dosažení terapeutické odpovědi ACR20 v týdnu 24. Sekundárními cíli byla odpověď ACR50 a ACR70 a změna DAS28 stanovená pomocí rychlosti sedimentace erytrocytů (erythrocyte sedimentation rate, DAS28-ESR) od počátku studie. Primární cíl byl splněn a terapeutická odpověď ACR20 byla splněna u SB5 v 72,4 % a u referenčního adalimumabu v 72,2 % (95 % CI -7,83 až 8,13 %) byla v rozmezí ekvivalence. Odpověď sledovaná v rámci sekundárních cílů byla v rozmezí ekvivalence ACR50: u SB5 byla 38,1 % a u referenčního adalimumabu 39,7 %, ACR70 pak u SB5 19,2 % a u referenčního adalimumabu 20,3 %. Nežádoucí účinky byly zaznamenány ve 35,8 % u SB5 a ve 40,7 % u referenčního adalimumabu, závažné nežádoucí účinky byly přítomny u 1,1 % a 2,9 % pacientů dané skupiny. Přítomnost ADA byla zaznamenána ve 33,1 % u SB5 a ve 32,0 % u referenčního infliximabu v týdnu 24. V týdnu 24 byli pacienti léčení referenčním adalimumabem znovu randomizováni podle podávané medikace: 129 pacientů pokračovalo v terapii referenčním adalimumabem, 125 pacientů bylo převedeno z referenčního adalimumabu na SB5 a 254 pacientů, kteří užívali SB5 od týdne 0, pokračovalo v terapii SB5. Terapeutická odpověď hodnocená podle ACR kritérií byla srovnatelná mezi jednotlivými rameny a udržela se po dobu 52 týdnů. Změna přípravku neměla vliv na účinnost či výskyt nežádoucích účinků mezi jednotlivými skupinami pacientů.41-43
FKB327 – Hulio
Biosimilární adalimumab FKB327 – Hulio je vyráběn firmou Mylan.33
Studie NCT02260791 (1. fáze) a NCT02405780 (2. fáze) byly dvojitě zaslepené randomizované studie fáze III porovnávající účinnost, bezpečnost a bioekvivalenci FKB327 (n = 367) s referenčním adalimumabem Humira (n = 363) u pacientů s aktivní RA, u kterých selhala terapie MTX. Primárním cílem bylo dosažení klinické odpovědi ACR20. Mezi sekundární cíle patřilo porovnání farmakologických vlastností, bezpečnosti a imunogenity. Primárního cíle bylo dosaženo s terapeutickou odpovědí ACR20 srovnatelnou u FKB327 (74,1 %; 95 % CI -7,9 až 4,7) s referenčním adalimumabem (75,7 %; 95 % CI -7,3 až 3,6), který byl v rámci stanovené hranice ekvivalence ± 13 %. U skóre DAS28-CRP byl rozdíl proti počátku u FKB327 3,43 a u referenčního infliximabu 3,42 (95 % CI -0,16 až 0,1). Podíl neutralizačních protilátek proti podávenému biologiku byl v týdnu 24 obdobný u FKB327 (57,7 %) a u referenčního adalimumabu (55,5 %). Studie byla rozdělena na dvě fáze s přelomovým 24. týdnem, kdy došlo k rerandomizaci. Do 2. fáze přešlo 645 pacientů, u kterých byl proveden switch medikace tímto způsobem: n = 216 FKB327^FKB327, n = 108 FKB327^referenční adalimumab (RP), n = 108 RP^FKB327, n = 213 RP^RP.
Po 30 týdnech užívali všichni pacienti FKB327 až do 76. týdne a byli sledováni další 4 týdny až do 80. týdne. Primárním koncovým ukazatelem v této studii byla bezpečnost, sekundárními koncovými ukazateli byla účinnost, přítomnost ADA a farmakologické vlastnosti. Ve 30. týdnu byla odpověď ACR20 srovnatelná u všech skupin a pohybovala se v rozmezí 71,3-75,5 % a 77,5-82,6 % pro skupiny FKB327^FKB327 a RP^RP. Odpověď ACR20 u pacientů se změnou léčby se pohybovala v rozmezí 67,6-82,4 % a 73,1-75,9 % pro skupiny FKB327^RP a RP^FKB327. Nebyly pozorovány žádné konzistentní rozdíly ve farmakologických vlastnostech a tvorbě neutralizačních protilátek a mezi jednotlivými skupinami. Biosimilární adalimumab FKB327 prokázal srovnatelnou bezpečnost a imunogenitu s referenčním adalimumabem.44,45
MSB11022 – Idacio
Přípravek MSB11022 – Idacio je biosimilární adalimumab firmy Fresenius Kabi.34
Studie AURIEL-RA byla randomizovaná dvojitě zaslepená studie fáze III porovnávající účinnost MSB11022 (n = 143) s referenčním adalimumabem (Humira) (n = 145) u pacientů se středně až závažně aktivní RA (n = 288). Cílem studie bylo prokázat bezpečnost a srovnatelnou účinnost obou přípravků. Terapeutické odpovědi hodnocené podle ACR20 dosáhlo ve 12. týdnu 79,6 % pacientů užívajících MSB11022 a 80,9 % pacientů s referenčním adalimumabem (95 % CI -10,55 až 8,04), tato odpověď na terapii byla konstantní po celou dobu studie do 52. týdne. Ostatní hodnocené parametry (ACR50/70, DAS28-ESR) byly rovněž konstantní u obou skupin. Přítomnost ADA byla zaznamenána u 80,4 % pacientů s MSB11022 a u 71,7 % pacientů užívajících referenční adalimumab, neutralizační protilátky pak 39,9 % a 39,3 % pro každé rameno. V 52. týdnu byla míra nežádoucích účinků zvláštního významu (hypersenzitivita) obdobná v obou skupinách: 4,2 % pro MSB11022 a 5,5 % pro referenční adalimumab. Výskyt nežádoucích příhod byl obdobný v obou skupinách nemocných (MSB11022 - 58,0 %; adalimumab RP - 64,1 %). Biosimilární adalimumab MSB11022 prokázal srovnatelnou účinnost a bezpečnost k referenčnímu adalimumabu.46
ABP 501 - Amgevita
Přípravek ABP 501 - Amgevita je biosimilární adalimumab vyvinutý firmou Amgen.35
Studie NCT01970475 byla randomizovaná dvojitě zaslepená studie fáze III hodnotila účinnost a bezpečnost ABP501 (n = 264) v porovnáním s referenčním adalimumabem (n = 262) u pacientů se středně těžkou až těžkou RA, u kterých došlo k selhání terapie MTX. Mezi cíle studie patřilo prokázat ekvivalenci obou studijních přípravků v rámci bezpečnosti, účinnosti a imunogenity. Ve. 24 týdnu dosáhlo terapeutické odpovědi hodnocené podle ACR20 74,6 % pacientů s ABP501 a 72,4 % pacientů s referenčním adalimumabem (relativní riziko [risk ratio] 1,039; 90 % CI 0,954-1,133), což splnilo požadavek předem definované ekvivalence. Průměr změny DAS28-CRP od počátku do 24. týdne hodnocení byl u obou skupin -2,32 (rozdíl mezi skupinami (0,01; 90 % CI -0,18 až 0,17). Nežádoucí příhody byly zaznamenané u 52,3 % pacientů, v jednotlivých skupinách pak u 50,0 % pacientů s ABP501 a u 54,6 % pacientů s referenčním adalimumabem. V otevřeném pokračování studie pokračovalo 229 pacientů s ABP501 a 237 pacientů s referenčním adalimumabem bylo převedeno na ABP501. Otevřená fáze nezaznamenala statisticky signifikantní rozdíly mezi terapeutickou odpovědí a mírou výskytu nežádoucích účinků, což bylo konzistentní s výsledky ze zaslepené fáze studie. Změna medikace neměla vliv na imunogenitu.47-49
CT-P17 – Yuflyma
Přípravek CT-P17 – Yuflyma j e biosimilární adalimumab firmy Celltrion.36
Studie NCT03789292 byla randomizovaná dvojitě zaslepená studie fáze III porovnávající CT-P17 (n = 324) s referenčním adalimumabem (n = 234) v populaci pacientů s aktivní RA se selháním předchozí terapie MTX. Hlavními cíli bylo posouzení účinnosti včetně radiografické progrese, farmakokinetika, bezpečnost a imunogenita. Odpověď ACR20 v týdnu 24 byla pro obě skupiny shodná 82,7 %. Ve 24. týdnu byla skupina s referenčním adalimumabem rerandomizována v poměru 1: 1, část pacientů užívala CT-P17 (n = 152) a část pokračovala s referenčním adalimumabem (n = 153). Pacienti užívající CT-P17 pokračovali s nezměněnou studijní medikací. V týdnu 52 dosáhlo terapeutické odpovědi ACR20 80,5 % pacientů, kteří byli od počátku na terapii CT-P17, 77,8 % pacientů pokračujících v terapii referenčním adalimumabem, při přechodu ve 24. týdnu na CT-P17 to bylo 82,2 %. Nebyl nalezen statisticky signifikantní rozdíl ve farmakologických vlastnostech, včetně imunogenity a výskytu nežádoucích účinků. Protilátky proti studijní medikaci byly detekovány u 28,4 % pacientů pokračujících s CT-P17, u 27,0 % pacientů pokračujících v referenčním adalimumabu, ve skupině s přechodem na CT-P17 v 24 týdnu byla zaznamenána pozitivita protilátek u 28,3 %.50,51
RITUXIMAB
Originální rituximab (Mabthera) je anti-CD20 protilátka využívaná v celé řadě oborů od hematoonkologie až po revmatologii, kde je většinou podáván nemocným se závažným průběhem choroby s orgánovým postižením, u kterého selhala předchozí konvenční terapie včetně bDMARD.
Charakteristika a dávkování rituximabu a jeho biosimilárních variant
Rituximab je chimérická monoklonální protilátka IgG1 složená z myší a lidské komponenty a namířená proti CD20. Předpokládaný mechanismu účinku rituximabu spočívá v lýze buněk CD20+ prostřednictvím vazby antigen vázajícího fragmentu (antigen-binding fragment, Fab) na CD20 těmito cestami: cytotoxickou (vyvolanou prostřednictvím komplementového systém) a protilátkově zprostředkovanou cytotoxicitou. Rituximab svojí vazbou na CD20 je schopen kromě výše zmíněných účinků vyvolat apoptózu B lymfocytů.
Dávkování u RA: Rituximab je indikován jako lék druhé volby po selhání alespoň jednoho blokátoru TNF- α. Ve výjimečných případech ho lze užít jako lék první volby tam, kde je terapie inhibitory TNF – α kontraindikována.1,2 Podává se obvykle intravenózně ve formě dvou infuzí v dávce 1 000 mg v odstupu 14 dnů. Již jsou dostupné práce hodnotící podávání polovičních dávek u RA s dostatečným účinkem na dosažení remise a jejího dlouhodobého udržení. Rituximab se podává i v dalších indikacích v revmatologii (tab. 3).52
GP2013 – Rixathon
Přípravek GP2013 – Rixathon je biosimilární rituximab firmy Sandoz.
Studie NCT01274182 byla randomizovaná dvojitě zaslepená studie s cílem prokázat farmarmakologickou ekvivalenci GP2013 a referenčního rituximabu (vyrobeného v EU nebo v USA) v kombinaci s MTX u pacientů s RA (n = 312) se selháním nebo intolerancí inhibitorů TNF- α. Ve 24. týdnu studie byly hodnoceny také další farmakologické parametry, včetně ekvivalence účinnosti a bezpečnosti. Geometrický průměr AUC byl v rozmezí předem dané ekvivalence 80-125 % pro všechna tři srovnání: GP2013 proti referenčnímu rituximabu EU (1,106; 90 % CI 1,010-1,210), GP2013 proti referenčnímu rituximabu USA (1,012; 90 % CI 0,925-1,108) a referenční rituximab EU proti referenčnímu rituximabu USA (1,093; 90% CI 0,989-1,208). Byla také prokázána ekvivalence deplece B buněk ve všech skupinách. Protilátky byly detekovány u 16,5 % ve skupině GP2013 oproti 15,1 % ve skupině s referenčním rituximabem až do poslední návštěvy pacienta ve studii. Odpověď ACR20 byla u GP2013 72,3 % (95 % CI 64,2-80,3) a u referenčního rituximabu 67,3 % (95 % CI 59,9-74,7) Účinnost, bezpečnost a profily imunogenity byly u GP2013 a referenčního rituximabu podobné.53 Další randomizovaná dvojitě zaslepená studie s paralelními skupinami se zaměřila na zhodnocení bezpečnosti přechodu z referenčního rituximabu na GP2013 ve srovnání s pokračováním terapie s referenčním rituximabem u pacientů s RA (n = 107). Nebyl nalezen statisticky významný rozdíl mezi přechodem na GP2013 či pokračováním v terapii referenčním rituximabem, výskyt hypersenzitivní reakce byla zaznamenán u 9,4 % u GP2013 a 11,1 % u referenčního rituximabu. Nebyly zachyceny neutralizační protilátky ani v jedné ze skupin. Při přechodu pacientů z referenčního rituximabu na GP2013 nebyla ve studii zaznamenána zvýšená bezpečnostní rizika.54
ÚČINNOST, BEZPEČNOST A IMUNOGENITA PŘECHODU MEZI BSDMARD A BSDMARD VYCHÁZEJÍCÍCH ZE STEJNÉHO ORIGINÁLNÍHO BDMARD
Dosud není vypracováno přesné doporučení pro přechod mezi bsDMARD a bsDMARD, což do jisté míry může představovat problém v rámci přechodu mezi jednotlivými přípravky. Klinické studie hodnotící změnu přípravku byly většinou zaměřeny na ovlivnění účinku změnou léku, bezpečnost a imunogenitu. Data národních registrů jsou rovněž cenným zdrojem údajů o změně přípravku z bsDMARD na bsDMARD. Data jsou zatím limitovaná charakterem studií, nicméně zdá se, že přechod z bsDMARD na bsDMARD není spojený se ztrátou účinnosti či zvýšením výskytu nežádoucích účinků.55
Infliximab - switch z CT-P13 na SB2 a CT-P13 na GP1111
Tříletá observační studie Laureta et al. hodnotila, zda switch z originálního infliximabu na biosimilární infliximab (CT-P13), po kterém následoval switch na druhý biosimilární infliximab (SB2), zvyšuje riziko imunogenity u pacientů s chronickým zánětlivým onemocněním (n = 140). Studie prokázala nízkou imunogenita (3 na 100 pacientoroků) bez jejího zvýšení při přechodu na jednotlivé bsDMARD.56
Neintervenční studie PERFUSE analyzovala 12měsíční data 496 pacientů s revmatickými chorobami (AS, RA a PsA), kteří užívali SB2. Studie prokázala dobré terapeutické a bezpečnostní vlastnosti SB2 jako prvního biologického léku, ale i jako druhého po switchi z referenčního infliximabu či jiného biosimilárního infliximabu. Celkem 75 % pacientů, kteří přešli z předchozího infliximabu, zůstalo na SB2 po 12 měsících od zahájení léčby SB2.57
Observační kohortová studie analyzovala celostátní přechod z originálního infliximabu na biosimilární CT-P13 a následně na biosimilární GP1111 u pacientů se zánětlivým revmatickým onemocněním (AS, RA, PsA) sledovaných v dánském registru DANBIO. V roce 2015 dokončilo první přechod celkem 780 pacientů; pacienti byli léčeni referenčním biologikem po dobu s mediánem sedm let. Celkem 83 % pacientů zůstalo v terapii
CT-P13 po prvním roce. V roce 2019 pokračovalo v léčbě 52 % pacientů léčených CT-P13 a byl proveden druhý switch na GP1111. Po jednom roce od přechodu z bsDMARD na bsDMARD zůstal GP1111 u 91 % pacientů. U přechodu bylo ukončení léčby spojeno s horšími výchozími ukazateli subjektivního hodnocení pacientem, ale objektivní ukazatele (CRP, skórovací škály aktivity) byly podobné.58
Adalimumab – switch z ABP 501 a SB5
Studie PERCEPTION se zabývala důsledky monoswitchingu a multiswitchingu v reálném prostředí klinické praxe z hlediska postojů pacientů k přechodu z jednoho biosimilárního adalimumabu na druhý. Pacienti (n = 90) s chronickými zánětlivými revmatickými onemocněními, včetně RA, AS a PsA byli rozděleni do skupin s monoswitchem (referenční adalimumab na biosimilární; n = 42) nebo multiswitchem (referenční biologikum na první biosimilární adalimumab a následně na druhý; n = 48). Studie ukázala, že multiswitching nevedl ke snížení spokojenosti pacientů, kteří užívali biosimilární lék. Zejména počet změn neměl na spokojenost pacientů žádný vliv.59
Etanercept – switch z GP2015 a SB4
Kiltzová et al. publikovali observační studii sledující celkem 100 pacientů (54 RA, 27 AS, 19 PsA), kteří přešli z biosimilárního etanerceptu SB4 na biosimilární etanercept GP2015 s průměrnou dobou sledování 21,1 ± 7,4 měsíce. Bylo zjištěno, že po několikanásobném switchi z originálního etanerceptu na SB4 a poté na GP2015 se míra retence blížila 90 % po šesti měsících po posledním switchi. V žádné indikaci nebyly zjištěny změny aktivity onemocnění po provedených změnách léku.60
ROLE BSDMARD V LÉČBĚ ZÁNĚTLIVÝCH REVMATICKÝCH ONEMOCNĚNÍ
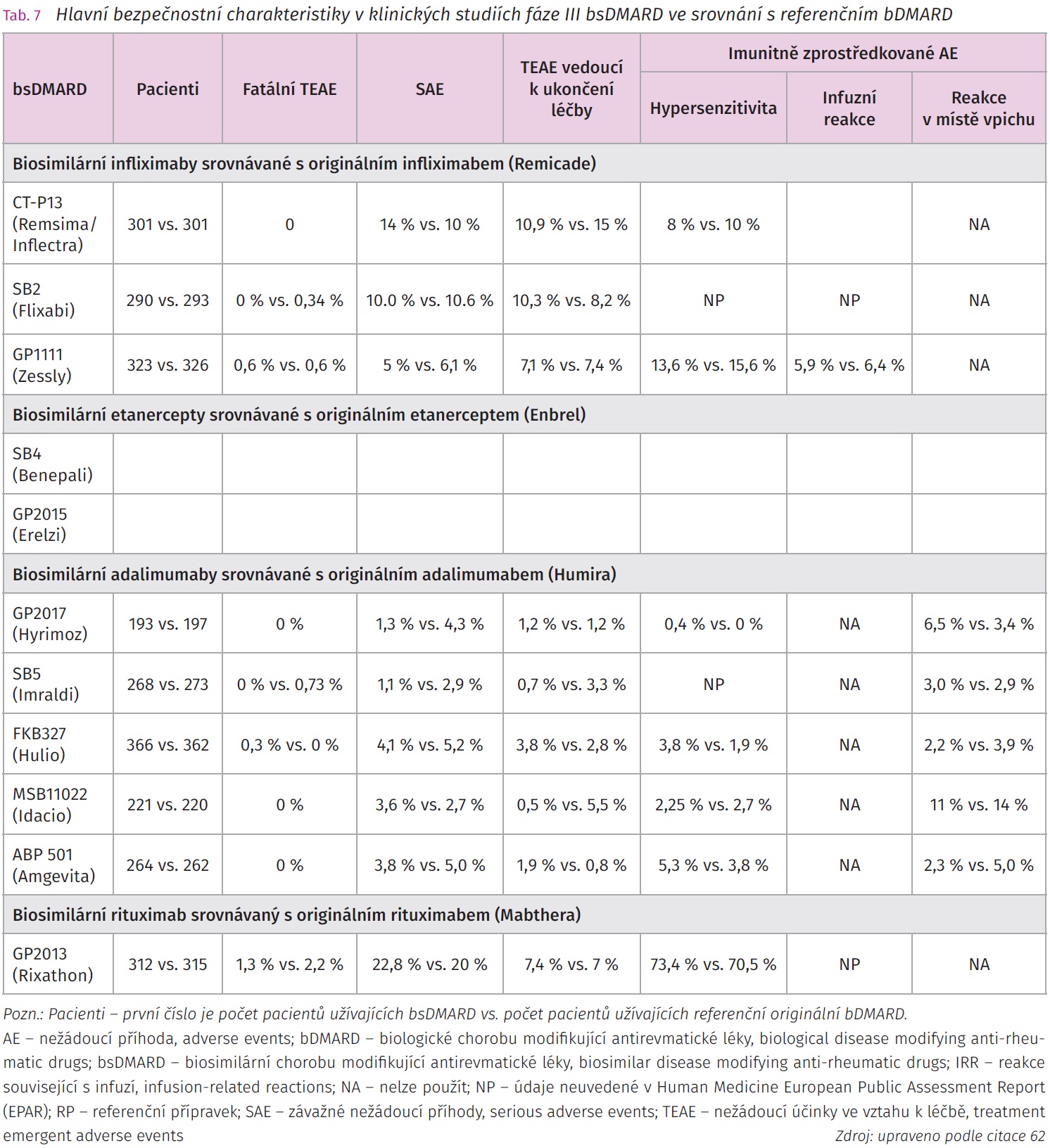
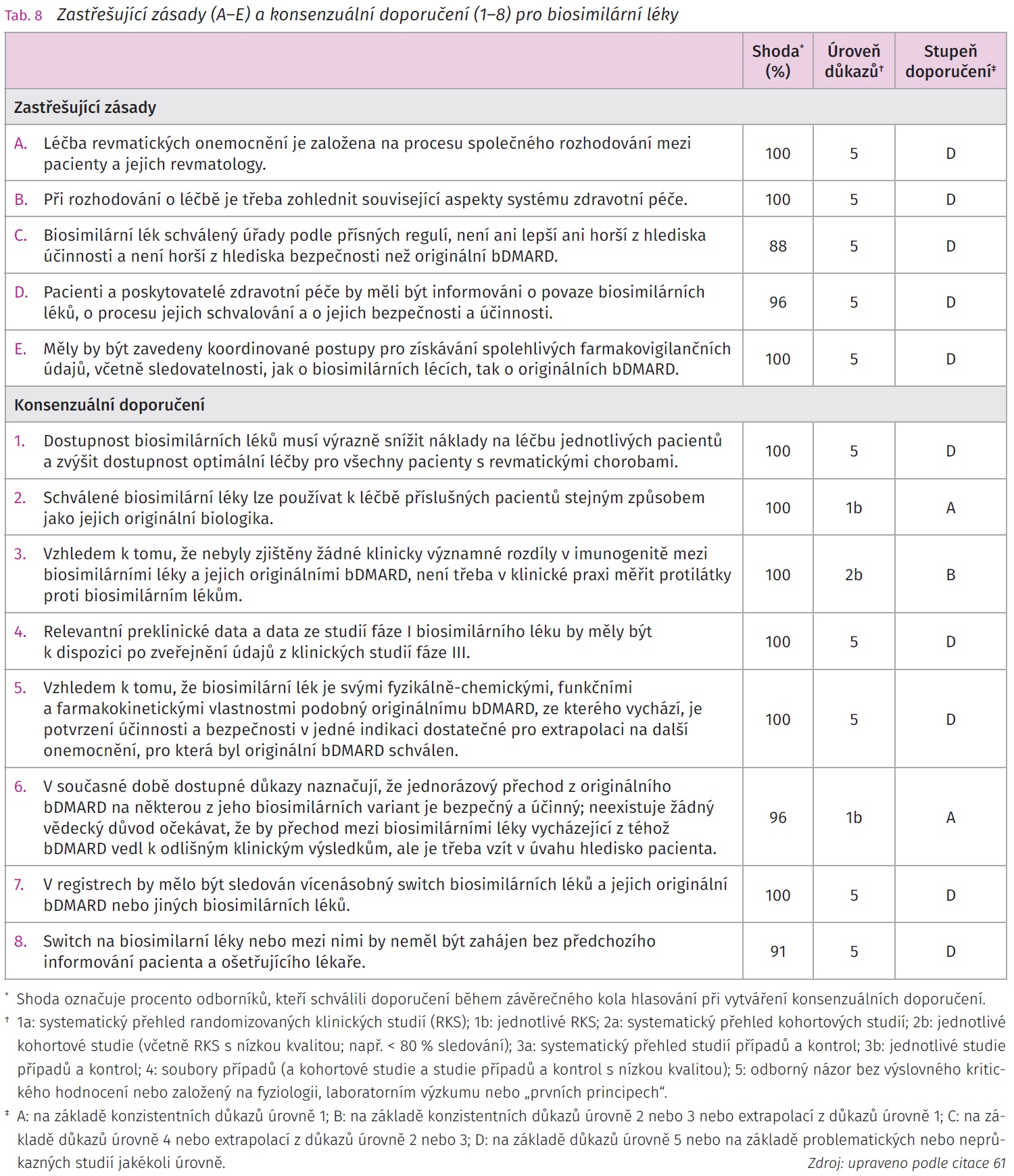
Biologická léčba výrazně zlepšila morbiditu a mortalitu pacientů se závažným průběhem chronických zánětlivých revmatických onemocnění. Náklady na léčbu představují výraznou zátěž zdravotnického systému a mohou se stát překážkou širšího rozšíření bDMARD v potřebné populaci pacientů, u kterých selhala předchozí konvenční terapie. V roce 2013 byl prvním schváleným bsDMARD pro klinické využití v Evropské unii biosimilární infliximab CT-P13 (Remsima/Inflectra). Další bsDMARD následovaly, v současnosti bylo schváleno na území EU více než 20 bsDMARD a lze předpokládat, že jejich počet se bude rozšiřovat. I přes dostupná data o bsDMARD přetrvává jistá rezervovanost k jejich použití v klinické praxi, obzvláště pak přechodu z originálního bDMARD na jeho bsDMARD variantu či při přechodu mezi jednotlivými bsDMARD. Velká studie NOR SWITCH byla zaměřena na sledování přechodu z referenčního bDMARD na bsDMARD v různých indikacích a neprokázala, že by změna měla vliv na účinnost, bezpečnost či imunogenitu pokračující léčby. Otevřené pokračovací observační fáze klinických sledování se potýkaly se zvýšenou mírou přerušení léčby po přechodu z referenčního biologika na biosimilární lék, toto bylo většinou připisováno více nocebo efektu než ztrátě účinnosti či vyššímu výskytu nežádoucích příhod. Další otázkou je vícenásobný přechod mezi referenčním bDMARD a dalšími bsDMARD (tab. 7).61,62 J. Kay et al. publikovali v roce 2018 pět zastřešujících zásad a osm konsenzuálních doporučení vypracovaných multidisciplinární skupinou, která analyzovala data získaná z klinických studií ohledně imunogenity, extrapolace indikací, switchi mezi originálním bDMARD a bsDMARD a mezi jednotlivými bsDMARD. Úroveň důkazů a stupeň doporučení se u každého z nich lišily podle dostupných publikovaných důkazů (tab. 8). Autoři doporučení poukazovali na nutnost dalších analýz přeregistračních a postmarketigových studií včetně dat z národních a nadnárodních registrů.61 Velká analýza dostupných regulačních dokumentů European Medicines Agency (EMA) publikovaná v roce 2021 se zabývala analýzou bezpečnosti, imunogenity a zaměnitelnosti biosimilárních protilátek (šest adalimumabů, tři infliximaby, tři rituximaby, dva bevacizumaby, šest trastuzumabů) a fúzních proteinů (tři etanercepty). Data prokázala, že četnost fatálních nežádoucích účinků, závažných nežádoucích účinků a imunitně podmíněných nežádoucích účinků byla srovnatelná mezi bsDMARD a jejich originálními bDMARD (tab. 7).62
Více než sedmiletá analýza postmarketigových dat neprokázala žádné nežádoucí účinky specifické pro bsDMARD. Dosud žádný bsDMARD nebyl stažen z klinické praxe z bezpečnostních důvodů, a to i přes narůstající počet pacientů užívajících bsDMARD jak naivních pro biologickou léčbu, tak převedených z originálního bDMARD na bsDMARD. Analýza údajů z klinických studií zabývajících se switchem z bDMARD/bsDMARD na bsDMARD v prokázala, že jednorázový nebo vícenásobný přechod mezi originálním přípravkem a jeho biosimilární verzí neměl žádný negativní dopad na účinnost, bezpečnost nebo imunogenitu.62 Nadnárodní doporučení EULAR, ACR, GRAPPA a z nich vycházející národní doporučení již zahrnují bsDMARD jako jednu z možných terapeutických voleb v léčbě pacientů s revmatickým onemocněním.6,7,63,64 Předpokládá se, že nižší náklady spojené s podáváním bsDMARD zvýší dostupnost biologické léčby většímu počtu pacientů.
ZÁVĚR
Biologická terapie přinesla v posledních několika dekádách revoluci v léčbě revmatických onemocnění a stala se nedílnou součástí celé řady terapeutických doporučení. Vypršení ochranných patentů originálních bDMARD otevřelo cestu k vývoji jejich biosimilárních variant, což přineslo celou řadu otázek, například zda je možná bezpečná změna bDMARD na jeho biosimilární variantu a naopak, či zda je možný switch bsDMARD na jiný bsDMARD vycházející ze stejného bDMARD, a další. Registrační studie některých bsDMARD byly navrženy tak, aby jejich data dokázala odpovědět na tyto otázky, což je u jednotlivých přípravků zmíněno. Výsledky registračních studií jednotlivých bsDMARD, postmarketingové studie a data z národních a nadnárodních registrů jsou zdrojem cenných dat stran bezpečnosti, účinnosti a imunogenity těchto přípravků. Podmínky registrace bsDMARD jsou přesně stanoveny regulačním lékovými agenturami, včetně pravidelné kontroly jejich kvality s důslednou farmakovigilancí. Nadnárodní americká a evropská doporučení pro léčbu revmatických onemocnění zahrnují bsDMARD jako jednu z možností volby biologického léku.
Podpořeno projektem koncepčního rozvoje výzkumné organizace MZ ČR 023728.
Literatura je k dispozici na www.farmakoterapeutickarevue.cz.